Abstract: Nanocrystalline HfB2 powders were successfully synthesized by molten salt synthesis technique at 1373 K using B and HfO2 as precursors within KCl/NaCl molten salts. The results showed that the as-synthesized powders exhibited an irregular polyhedral morphology with the average particle size of 155 nm and possessed a single-crystalline structure. From a fundamental aspect, we demonstrated the molten-salt assisted formation mechanism that the molten salts could accelerate the diffusion rate of the reactants and improve the chemical reaction rate of the reactants in the system to induce the synthesis of the high-purity nanocrystalline powders. Thermogravimetric analysis showed that the oxidation of the as-synthesized HfB2 powders at 773–1073 K in air was the weight gain process and the corresponding oxidation behavior followed parabolic kinetics governed by the diffusion of oxygen in the oxide layer.
Keywords: ultra-high temperature ceramics; powders; molten salt synthesis; oxidation behavior
1 Introduction
As members of ultra-high temperature ceramics, transition-metal borides (TMB2) ceramics have attracted considerable attentions for potential applications in aircraft, atomic, and astronautic manufacturing industries, since they exhibit high melting point, high hardness, high thermal conductivity, and excellent chemical stability [1–3]. The synthesis of TMB2 powders is critical for implementing their extensive applications. Up to now, a variety of techniques have been developed to synthesize TMB2 powders, such as mechanochemically assisted method [4–7], carbo/borothermal reduction [8–10], sol–gel method [11–13], and so on. However, the reported techniques for synthesizing TMB2 powders have some shortcomings, such as the high synthesis temperatures and expensive reaction precursors. In addition, the as-obtained powders possess the large
particle size. Therefore, a low temperature technique for synthesizing TMB2 ultrafine powders is of vital importance for implementing the extensive applications of TMB2 ceramics. Recently, a low temperature molten salt synthesis technique has been proposed to synthesize TMB2 ultrafine powders including NbB2 [14], TiB2 [15], and CrB2 [16]. Nevertheless, the current understanding of the molten-salt assisted formation mechanism is yet limited, and the synthesis of much more TMB2 ultrafine powders by the molten salt synthesis technique has rarely been reported until now. In addition, there is seldom report so far on the oxidation behavior of TMB2 nanopowders, which is a critical property for high-temperature applications.
HfB2 ceramics, as a member of TMB2 ceramics, have recently attracted extensive attentions because they exhibit potential applications in high-temperature electrodes and thermal protection systems for hypersonic aerospace vehicles [17–20]. In this work, we successfully synthesized the nanocrystalline HfB2 powders by the molten salt synthesis technique at a relatively low temperature of 1373 K using the inexpensive HfO2 and B powders as reaction precursors within the inexpensive KCl/NaCl molten salts. The effect of synthesis temperatures and molten salts on the phase compositions, morphology, and microstructure of the as-synthesized powders was investigated in detail, as well as the molten-salt assisted formation mechanism. The oxidation behavior of the as-synthesized powders was also investigated at different temperatures.
2 Experimental
The synthesis of HfB2 powders was conducted by the molten-salt assisted borothermal reduction based on the reaction between HfO2 powders (99.9% purity, average particle size: 1–3 μm, Shanghai Chaowei Nanotechnology Co., Ltd., Shanghai, China) and amorphous B powders (purity: 99.9%, average particle size < 3 μm, Shanghai Chaowei Nanotechnology Co., Ltd., Shanghai, China) with the presence of NaCl/KCl molten salts (molar ratio: 1:1, eutectic point: 931 K). The molar ratio was 3:10 for HfO2/B and 1:1 for NaCl/KCl, respectively. The weight ratio was 10:1 for NaCl/KCl and HfO2/B. Details of the synthesis of HfB2 powders were described as follows: The starting materials of HfO2, B, and NaCl/KCl were first mixed by hand for 30 min in an agate mortal using an agate pastel, and then the mixture of raw materials was put into an alumina crucible and placed into the horizontal alumina tube furnace. After evacuating the furnace for three times, argon carrier gas (purity: 99.99%) was introduced into the system with a flow rate of 300 sccm (standard-state cubic centimeter per minute). The system was heated from room temperature to 1273–1373 K at a rate of 10 K/min and held for 1 h, followed by furnace cooling to room temperature. Afterwards the as-synthesized products were taken out and immersed in the deionized water at 353 K to dissolve the residual NaCl/KCl salts, and B2O3 production. Finally, they were filtered and washed by the deionized water and the absolute ethanol for several times, and dried at 333 K. The products were also prepared by the similar borothermal reduction method without NaCl/KCl molten salts for comparison.
Thermogravimetric analysis (TGA) of the as-synthesized powders was performed using a Metter Toledo Star TGA/SDTA 851 thermal analyzer to investigate their isothermal oxidation behaviors at different temperatures. Before the isothermal oxidation tests, the surface area of the as-synthesized HfB2 powders was first measured to be about 3.5 m²·g-1 by Brunauer–emmett–teller Test method. Afterwards the samples were heated to the desired temperatures (773, 873, 973, and 1073 K) for the isothermal oxidation tests in air. The fast heating stage with a heating rate of 50 K/min prior to the isothermal period was applied to minimize oxidation effects before reaching the target temperatures in flowing nitrogen (purity: 99.99%) with a flow rate of 50 sccm. Finally, they were cooled naturally to room temperature in flowing nitrogen with a flow rate of 50 sccm. Weight changes of the as-synthesized powders related to the oxidation time were recorded with thermogravimetric mode.
The samples were analyzed by X-ray diffraction (XRD, X’pert PRO; PANalytical, Almelo, the Netherlands), scanning electron microscopy (SEM, Supra-55; Zeiss, Oberkochen, Germany), and transmission electron microscopy (TEM, Tecnai F30G2; FEI, Eindhoven, the Netherlands) equipped with energy dispersive spectroscopy (EDS).
3 Results and discussion
XRD patterns of the as-synthesized products at various conditions are shown in Fig. 1. Clearly, after incorporating molten salts, it can be seen that the products synthesized at 1273 K consist of a dominant HfB2 phase and a minor HfO2 impurity phase, as displayed in Fig. 1(a). However, when the synthesis temperature rises to 1373 K, the diffraction peaks of HfO2 impurity phase disappear and only the diffraction peaks of HfB2 phase can be detected in the as-synthesized products, as shown in Fig. 1(b). This indicates that HfB2 products can be successfully synthesized at 1373 K with molten salts. Before incorporating molten salts, the products synthesized at 1373 K are composed of HfB2 phase and HfO2 impurity phase, as presented in Fig. 1(c). Obviously, a high content of HfO2 impurity phase is present in the as-synthesized products. This suggests that HfB2 products cannot be synthesized at 1373 K without molten salts, which clearly demonstrates that the presence of molten salts induces the synthesis of HfB2 products.
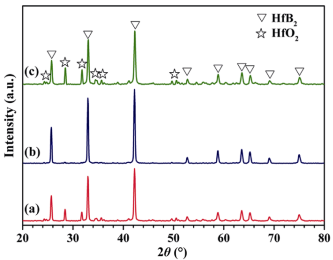
Fig. 1 XRD patterns of the as-synthesized products at different conditions: (a) 1273 K with molten salts, (b) 1373 K with molten salts, and (c) 1373 K without molten salts.
SEM images of HfB2 products synthesized at 1373 K with molten salts are displayed in Fig. 2. From Fig. 2(a),it is evident that the as-synthesized products consist of a large amount of small particles. The high magnification SEM image shows that these small particles possess an irregular polyhedral morphology with the relatively uniform particle sizes, as shown in Fig. 2(b). In addition, SEM image can be examined to determine their particle sizes. A total of 100 individual particles were measured to get the average value of particle sizes and a Gaussian fitting to these data yields an average particle size of 155 nm (Fig. 2(c)). Therefore, the nanocrystalline HfB2 powders can be successfully synthesized at 1373 K with molten salts.

Fig. 2 SEM characterizations of HfB2 products synthesized at 1373 K with molten salts: (a) low magnification SEM image and (b) high magnification SEM image. (c) Histogram of each measured data for the particle sizes with a Gaussian fitting to the data. The Gaussian peak is centered at 155 nm.
Figure 3(a) shows a typical TEM image of HfB2 powders synthesized at 1373 K with molten salts, from which it can be clearly observed that the as-synthesized powders involve a large number of individual nanocrystalline particles. Clearly, their particle sizes can also be determined based on TEM images. A total of 50 individual particles were measured to get the average value of particle sizes. A Gaussian fitting to these data yields an average particle size of 155 nm, as shown in the inserted histogram in Fig. 3(a), which is in good agreement with the measured results from SEM images (Fig. 2(c)). Figure 3(b) shows a representative selected area electron diffraction (SAED) pattern along zone axis [111] of the as-synthesized powders. It clearly exhibits that the as-synthesized powders are the single crystal and hexagonal structure of HfB2 due to the well-arranged diffraction spots with the symmetry. The high-resolution transmission electron microscopy (HRTEM) image of the as-synthesized powders in Fig. 3(c) shows a periodic lattice structure and a set of fringes with the d-spaces of 0.272 nm, corresponding to the (110) plane of HfB2 (JCPSD Card No. 12-0234), which further confirms that the as-synthesized powders are single-crystalline hexagonal HfB2. In addition, an obvious amorphous B2O3 layer of ~4 nm can be seen on each particle surface (marked by dotted red lines in Fig. 3(c)), which could account for the presence of O element in EDS spectrum, as displayed in Fig. 4(d). The scanning transmission electron microscopy (STEM)–EDS analysis was further performed at an acceleration voltage of 200 kV and a collection time of 230 s. Besides O element, EDS spectrum also reveals that the as-synthesized powders contain Hf, B, and Cu elements. The presence of Hf and B elements verifies that the as-synthesized powders are composed of HfB2 phases. The detected Cu element should be attributed to the copper grids used to support TEM samples.
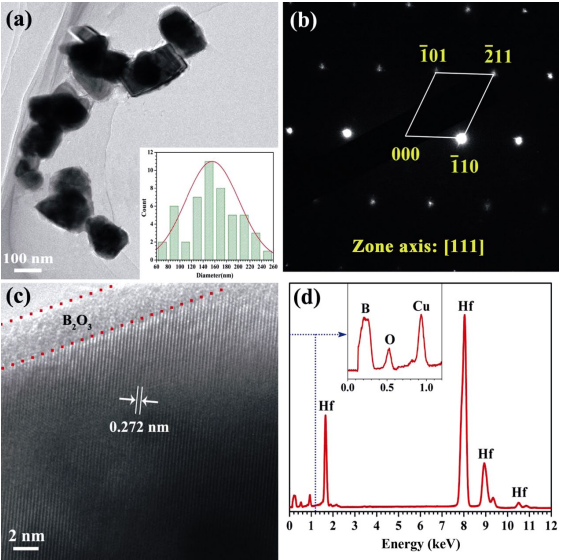
Fig. 3 TEM analysis of HfB2 products synthesized at 1373 K with molten salts: (a) TEM image, (b) SAED pattern, (c) HRTEM image, and (d) EDS spectrum.
On the basis of the aforementioned experiment results, it can be concluded that the presence of molten salts is crucial for the synthesis of the nanocrystalline HfB2 powders in our work. Similar conclusions have also been reported previously in other TMB2 ultrafine powders synthesized by the molten-salt assisted method, such as NbB2 [14], TiB2 [15], and CrB2 [16]. However, the molten-salt assisted formation mechanismis not well understood. To better understand this mechanism, we establish two simple schematic diagrams of formation mechanisms associated to the as-synthesized HfB2 powders without molten salts and with molten salts, as shown in Fig. 4.
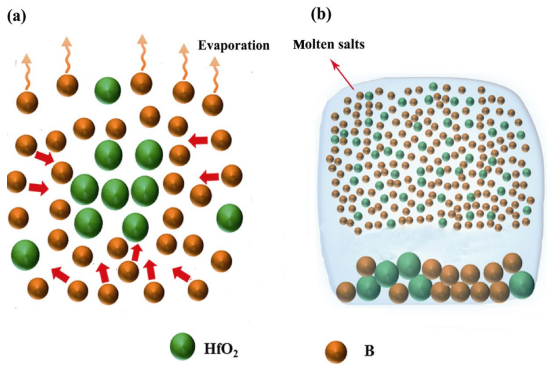
Fig. 4 Schematic diagrams of formation mechanisms associated to the as-synthesized HfB2 powders: (a) without molten salts and (b) with molten salts.
In our case, the reaction precursors of the system involve HfO2 powders and amorphous B powders. NaCl/KCl is only used as molten salts. As a result, before incorporating molten salts, the synthesis of HfB2 powders is a traditional solid–solid reaction process, in which B reactant will first diffuse to the surface of HfO2 reactant and then it reacts with HfO2 to generate HfB2 product at synthesis temperatures, as displayed in Fig. 4(a). The reaction as well as the correlation between the standard Gibbs free energy (∆GθR,T ) of this reaction and the oxidation temperature (T) can be described as follows [21]:
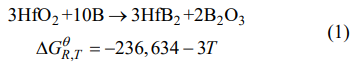
where R is the gas constant. It is evident that the standard Gibbs free energy of Reaction (1) is so negative that this reaction can occur spontaneously at synthesis temperatures. After incorporating molten salts, the synthesis of HfB2 powders is a new molten-salt assisted liquid–solid reaction process, as presented in Fig. 4(b). In this process, the molten salts will be first melted into isotropic liquid at elevated temperatures, and then the B and HfO2 reactants are rapidly dissolved into the molten salts. This finally leads to the uniformly distributions of B and HfO2 in the molten salts. Afterwards HfB2 product is generated by Reaction (1) between the B and HfO2 reactants at synthesis temperatures by Eq. (1). With the reaction continuing, the dissolved reactants are constantly consumed, but at the same time the undissolved reactants are continuously dissolved into molten salts to participate in the reaction until the end. In general, the total reaction rate (V) of the system depends on the diffusion rate (VD) of the reactants and the chemical reaction rate (VR) of the reactants, which can be calculated by the following equations [22]:
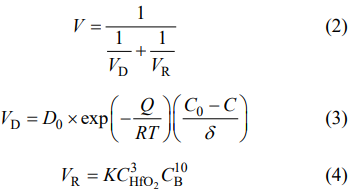
where Q is the diffusion activation energy, D0 is the constant of coefficient of diffusion, C0 is the reactant concentration at the beginning of diffusion, C is the reactant concentration at the end of diffusion, δ is the diffusion distance, K is the chemical reaction constant, CHfO2 and CB are the HfO2 and B concentrations, respectively. From Eq. (3), it can be found that the diffusion rate of the reactants is closely related to the diffusion distance and the diffusion activation energy. Before incorporating molten salts, there is a long diffusion distance in the reactants due to their agglomerations, as shown in Fig. 4(a). After incorporating molten salts, the B and HfO2 reactants are uniformly dispersed into the molten salts, and thereby a short diffusion distance is presented in the reactants, as displayed in Fig. 4(b). In addition, as a rule, the diffusion activation energy of the reactants in the liquid phase is much less than that in the solid phase. As a consequence, both the short diffusion distance and the reduced diffusion activation
energy result in the presence of a large diffusion rate in the molten-salt assisted synthesis process. From Eq. (4), we can see that the chemical reaction rate of the reactants is mainly determined by the HfO2 and B concentrations. In general, B reactant is very easy to be volatilized at elevated temperatures. However, the degree of B evaporation in liquid phase is less than that in solid phase. Consequently, B reactant possesses a high concentration in the molten-salt assisted synthesis process, which could significantly enhance the chemical reaction rate of the reactants. In brief, the presence of molten salts not only accelerates the diffusion rate of the reactants by shortening their diffusion distances and decreasing their diffusion activation energies, but also enhances the chemical reaction rate by reducing the evaporation of B. Owing to the high diffusion rate
and chemical reaction rate, according to Eq. (2), it can be clearly observed that the total reaction rate of the system will be high in the molten-salt assisted synthesis process, which finally results in the synthesis of the nanocrystalline HfB2 powders.
To investigate the isothermal oxidation behavior of the as-synthesized HfB2 powders at different temperatures, the TGA test was conducted and the results are shown in Fig. 5.
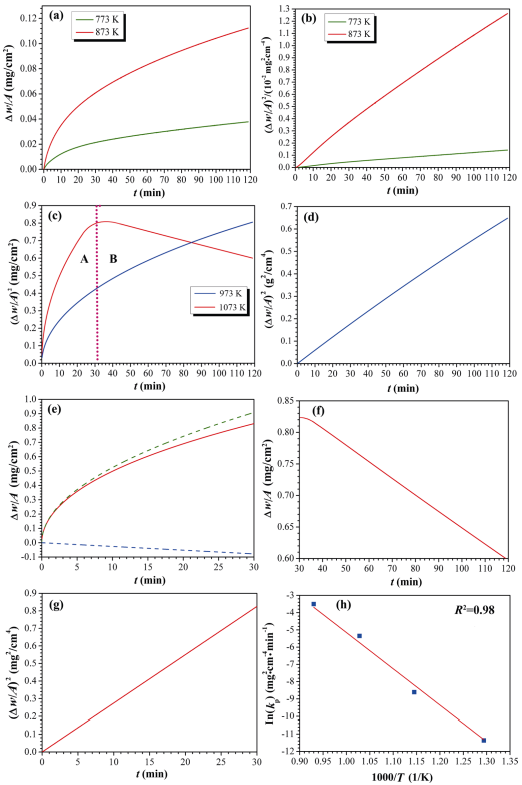
Fig. 5 Oxidation analysis of the as-synthesized HfB2 powders by TGA tests at different temperatures: (a) weight change per unit area (∆w/A, ∆w is the weight change and A is the surface area of samples) as a function of oxidation time (t) at 773 and 873 K; (b) square of the weight change per unit surface area as a function of oxidation time at 773 and 873 K; (c) weight change per unit area as a function of oxidation time at 973 and 1073 K; (d) square of the weight change per unit surface area as a function of oxidation time at 973 K; (e) weight change per unit area as a function of oxidation time at 1073 K after 30 min; (f) weight change per unit area as a function of oxidation time at 1073 K before 30 min with fitted parabolic and line curves; (g) square of the weight change per unit surface area corresponding to the fitted parabolic curve in (f); (h) plot of logarithm parabolic rate kp (parabolic rate constant) versus reciprocal of the temperatures.
From Fig. 5(a), it can be seen that the oxidation of the samples at 773 and 873 K exhibits the weight gain process. However, the oxidation rate of the samples at 873 K is much higher than that of the samples at 773 K. After isothermal oxidation for 120 min in air, the weight gain of the samples at 773 K is only 0.034 mg/cm², while the weight gain of the samples at 873 K is up to 0.116 mg/cm². Figure 5(b) depicts the plots of the square of the specific weight change as a function of oxidation time for the samples at 773 and 873 K. Clearly, there is a good linear relationship (R² > 0.99) between the square of the specific weight change and oxidation time, which indicates that the oxidation behavior of the samples at 773 and 873 K follows the parabolic kinetics. That is to say, the oxidation of the samples at 773 and 873 K is governed by a diffusion process that oxygen diffused into the oxide layer generated in the reaction between HfB2 and O2 by the following equation [23,24]:
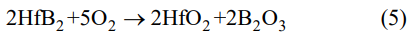
As the oxidation temperature rises to 973 K, the oxidation of the samples is still a weight gain process and their oxidation behavior also follows a parabolic kinetics (Figs. 5(c) and 5(d)). However, the weight gain of the samples is up to 0.8 mg/cm2 after isothermal oxidation for 120 min in air, much larger than that of the samples at 873 K, suggesting that the oxidation rate of the samples at 973 K is quite high. When the oxidation temperature increases to 1073 K, the oxidation of the samples is a complicated weight gain process, as displayed in Fig. 5(c), which can be divided into two stages, marked as A and B, respectively. During the stage A (0–30 min), the oxidation of the samples exhibits a rapid weight gain and the final weight gain is up to 0.82 mg/cm2 after isothermal oxidation for 30 min in air. As the oxidation time is over 30 min (stage B), the oxidation of the samples is still a weight gain process, but the weight gain rate is negative. Hence, the final weight gain decreases to 0.6 mg/cm² after isothermal oxidation for 120 min in air. In addition, From Eq. (5), it can be found that the oxidation of the samples is a weight gain process. Therefore, it can be concluded that the samples have been completely oxidized after isothermal oxidation for 30 min at 1073 K in air. After isothermal oxidation for 30 min, the weight gain of the samples is a linear decrease process (Fig. 5(f)), which should be associated to the evaporation of B2O3. On the basis of this linear relationship, a constant evaporation rate of B2O3 can be calculated to be ~0.026 mg/(cm²∙min-1). Before isothermal oxidation for 30 min, it should be noted that the oxidation behavior of the samples deviates from the parabolic kinetics, as shown in Fig. 5(e). Therefore, a simple parabolic law is unable to describe the oxidation kinetic behavior. However, the TGA curve can be fitted into a linear term and a parabolic term based on a multiple-law model, according to the following equation [25]:
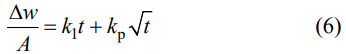
where kl is the linear rate constant. The results are depicted in Fig. 5(e). Obviously, the dominating term is still a parabolic curve (green dotted line), which suggests that the oxidation behavior of the samples is still a parabolic kinetics before isothermal oxidation for 30 min at 1073 K in air. This can be further confirmed by the presence of a good linear relationship between the square of the specific weight change and oxidation time, as depicted in Fig. 5(g). The rest term is a linear curve (blue dotted line) and it is negative, which is associated with the evaporation of B2O3. This is because that the slop of the line can be calculated to be ~0.025 mg/(cm²∙min-1), which is consistent with the vaporization rate of B2O3 (0.026 mg/(cm²∙min-1)) after isothermal oxidation for 30 min. In sum, the oxidation behavior of the samples follows the parabolic kinetics at 773–1073 K, which is governed by the diffusion of oxygen in the oxide layer. As a result, kp at various oxidation temperatures can be calculated by the following equation [26]:

The calculated results are listed in Table 1. On the basis of these results, for the oxygen diffusion mode, the oxidation activation energy (∆E) of the samples can be also calculated by the following equation [27]:
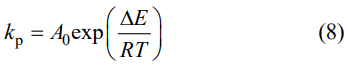
where A0 is the constant of preexponential factor. Figure 5(h) plots the In(kp) as a function of reciprocal temperatures, and thereby the oxidation activation energy can be calculated to be ~190.8 kJ/mol.
Table 1 Calculated kp with corresponding R² (fit goodness) for various oxidation temperatures
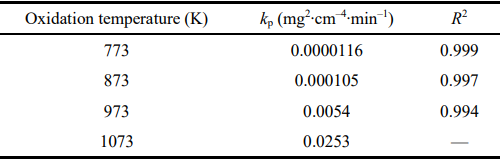
Figure 6 shows that XRD patterns of the as-synthesized HfB2 powders after isothermal oxidation tests at various temperatures. After isothermal oxidation tests at 773 and 873 K, the as-synthesized HfB2 powders are mainly composed of a dominant HfB2 phase and a minor HfO2 phase, as displayed in Fig. 6(a), which suggests that only a small number of HfB2 powders are oxidized at 773 and 873 K. As the oxidation temperature rises to 973 K, the as-synthesized HfB2 powders mainly consist of a dominant HfO2 phase and a minor HfB2 phase, indicating that most of HfB2 powders have been oxidized at 973 K. When the oxidation temperature increases to 1073 K, the diffraction peaks of HfB2 phase have completely disappeared in the XRD pattern and all the relatively sharp diffraction peaks in the XRD pattern can be indexed to HfO2 phase. These imply that HfB2 powders have been completely oxidized at 1073 K. What’s more, the amorphous B2O3 can also be detected by XRD, as depicted in Fig. 6(b). A weak diffraction peak near a 2θ value of 28° from the {310} lattice plane of the amorphous B2O3 product can be observed at 773 and 873 K, while its intensity becomes very remarkable when the oxidation temperatures rise to 973 and 1073 K. This further confirms that only a small number of HfB2 powders are oxidized at 773 and 873 K, while most or all of HfB2 powders have been oxidized at 973 and 1073 K.
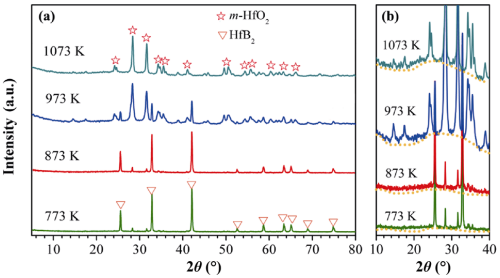
Fig. 6 XRD analysis of the as-synthesized HfB2 powders after oxidation tests: (a) XRD patterns of the as-synthesized HfB2 powders after oxidation tests at various temperatures;(b) high magnification of XRD patterns with 2θ from 10° to 40°.
Figure 7 shows that the SEM images of the as-synthesized HfB2 powders after isothermal oxidation tests at various temperatures. After isothermal oxidation tests at 773 and 873 K, the powders mainly consist of the irregularly HfB2 particles, on which some amorphous B2O3 glass and HfO2 nanoparticles can be found, as shown in Figs. 7(a) and 7(b). This suggests that the powders are only oxidized slightly at 773 and 873 K. As the oxidation temperature rises to 973 K, the powders are mainly composed of numerous HfO2 nanoparticles and amorphous B2O3 glass, and only a few irregularly HfB2 particles can be observed, indicating that most of HfB2 powders have been oxidized at 973 K. When the oxidation temperature increases to 1073 K, the powders involve numerous HfO2 nanoparticles, nanorods, and amorphous B2O3 glass phase can be found, which implies that all of HfB2 powders have been completely oxidized at 973 K.

Fig. 7 SEM images of the as-synthesized HfB2 powders after oxidation tests at various temperatures in air: (a) 773, (b) 873, (c) 973, and (d) 1073 K.
4 Conclusions
In summary, the nanocrystalline HfB2 powders had been successfully synthesized via a simple molten salt synthesis technique. The as-synthesized HfB2 powders possessed an irregular polyhedral morphology with the average particle sizes of ~155 nm. In addition, they also exhibited a single-crystalline structural feature. More importantly, from a fundamental aspect, we demonstrated why the presence of molten salts could induce the synthesis of nanocrystalline HfB2 powders. The oxidation results show that the oxidation of as-synthesized HfB2 powders at 773–1073 K in air is the weight gain process and the corresponding oxidation behavior follows parabolic kinetics that is governed by the diffusion of oxygen in the oxide layer. This work not only may provide a new method for the synthesis of HfB2 powders and a theoretical guidance for the molten salt synthesis method, but also reveal the oxidation behaviors of the nanocrystalline HfB2 powders at low temperatures in air.
References
[1] Fahrenholtz WG, Hilmas GE, Talmy IG, et al. Refractory diborides of zirconium and hafnium. J Am Ceram Soc 2007, 90: 1347–1364.
[2] Gui KX, Liu FY, Wang G, et al. Microstructural evolution and performance of carbon fiber-toughened ZrB2 ceramics with SiC or ZrSi2 additive. J Adv Ceram 2018, 7: 343–351.
[3] Ren XR, Feng PZ, Guo LT, et al. Synthesis of ultra-fine TaB2 nano powders by liquid phase method. J Am Ceram Soc 2017, 100: 5358–5362.
[4] Guo SQ, Hu CF, Kagawa Y. Mechanochemical processing of nanocrystalline zirconium diboride powder. J Am Ceram Soc 2011, 94: 3643–3647.
[5] Radev DD, Klissurski D. Mechanochemical synthesis and SHS of diborides of titanium and zirconium. J Mater Synth Process 2001, 9: 131–136.
[6] Guo SQ, Ping DH, Kagawa Y. Synthesis of zirconium diboride platelets from mechanically activated ZrCl4 and B powder mixture. Ceram Int 2012, 38: 5195–5200.
[7] Chen LY, Gu YL, Shi L, et al. Synthesis and oxidation of nanocrystalline HfB2. J Alloys Compd 2004, 368: 353–356.
[8] Guo WM, Zhang GJ, You Y, et al. TiB2 powders synthesis by borothermal reduction in TiO2 under vacuum. J Am Ceram Soc 2014, 97: 1359–1362.
[9] Ni DW, Zhang GJ, Kan YM, et al. Synthesis of monodispersed fine hafnium diboride powders using carbo/borothermal reduction of hafnium dioxide. J Am Ceram Soc 2008, 91: 2709–2712.
[10] Ma L, Yu JC, Guo X, et al. Effects of HBO2 on phase and morphology of ZrB2 powders synthesized by carbothermal reduction. Ceram Int 2017, 43: 12975–12978.
[11] Yang BY, Li JP, Zhao B, et al. Synthesis of hexagonal-prism-like ZrB2 by a sol-gel route. Powder Technol 2014, 256: 522–528.
[12] Rabiezadeh A, Hadian AM, Ataie A. Synthesis and sintering of TiB2 nanoparticles. Ceram Int 2014, 40: 15775–15782.
[13] Patra N, Nasiri NA, Jayaseelan DD, et al. Synthesis, characterization and use of synthesized fine zirconium diboride as an additive for densification of commercial zirconium diboride powder. Ceram Int 2016, 42: 9565–9570.
[14] Ran SL, Sun HF, Wei YN, et al. Low-temperature synthesis of nanocrystalline NbB2 Powders by borothermal reduction in molten salt. J Am Ceram Soc 2014, 97: 3384–3387.
[15] Bao K, Wen Y, Khangkhamano M, et al. Low-temperature preparation of titanium diboride fine powder via magnesiothermic reduction in molten salt. J Am Ceram Soc 2017, 100: 2266–2272.
[16] Liu ZT, Wei YN, Meng X, et al. Synthesis of CrB2 powders at 800 ℃ under ambient pressure. Ceram Int 2017, 43: 1628–1631.
[17] Zhang GJ, Ni DW, Zou J, et al. Inherent anisotropy in transition metal diborides and microstructure/property tailoring in ultra-high temperature ceramics—A review. J Eur Ceram Soc 2018, 38: 371–389.
[18] Jalaly M, Gotor FJ, Sayagués MJ. Self-propagating mechanosynthesis of HfB2 nanoparticles by a magnesiothermic reaction. J Am Ceram Soc 2018, 101: 1412–1419.
[19] Ren XR, Shang TQ, Wang WH, et al. Dynamic oxidation protective behaviors and mechanisms of HfB2-20wt%SiC composite coating for carbon materials. J Eur Ceram Soc 2019, 39: 1955–1964.
[20] Liang H, Guan SX, Li X, et al. Microstructure evolution, densification behavior and mechanical properties of nano-HfB2 sintered under high pressure. Ceram Int 2019, 45: 7885–7893.
[21] Fahrenholtz WG, Binner J, Zou J. Synthesis of ultra-refractory transition metal diboride compounds. J Mater Res 2016, 31: 2757–2772.
[22] Toyoura K, Koyama Y, Kuwabara A, et al. First-principles approach to chemical diffusion of lithium atoms in a graphite intercalation compound. Phys Rev B 2008, 78: 214303.
[23] Parthasarathy TA, Rapp RA, Opeka M, et al. A model for the oxidation of ZrB2, HfB2 and TiB2. Acta Mater 2007, 55: 5999–6010.
[24] Zapata-Solvas E, Jayaseelan DD, Brown PM, et al. Effect of La2O3 addition on long-term oxidation kinetics of ZrB2-SiC and HfB2-SiC ultra-high temperature ceramics. J Eur Ceram Soc 2014, 34: 3535–3548.
[25] Nickel KG. Corrosion of Advanced Ceramics: Measurement and Modelling. Dordrecht, the Netherlands: Kluwer Academic Publishers, 1994.
[26] Harrison RW, Lee WE. Mechanism and kinetics of oxidation of ZrN ceramics. J Am Ceram Soc 2015, 98: 2205–2213.
[27] Deal BE, Grove AS. General relationship for the thermal oxidation of silicon. J Appl Phys 1965, 36: 3770–3778.
Declaration: This article is provided by CERADIR™ users or obtained from Internet, the content does not represent the position of CERADIR™. We are not responsible for the authenticity/accuracy of the article, especially the effects of the products concerned. This article is for study only, it does not constitute any investment or application advice. For reprinting, please contact the original author. If it involves the copyright and/or other issues, please contact us and we will deal with it asap! CERADIR™ has the interpretation of this declaration.