摘要: 为改善钛酸铋钠基无铅陶瓷的铁电光伏特性,通过传统固相法制备了B位Mo掺杂的Na0.5Bi0.5(Ti1-xMox)O3(BNT-Mox,x=0~0.02)无铅铁电陶瓷。通过XRD、拉曼光谱、吸收光谱等测试方法,结合基于密度泛函理论的第一性原理计算,研究了Mo掺杂对BNT陶瓷体系带隙的影响规律及机理。结果表明:随着Mo掺杂量的增加,光学带隙值先减小后增大,当x=1.0%时带隙达到最小值2.33 eV,并且光吸收强度达到最大值69%;通过对能带和态密度计算结果进行分析,发现Mo掺杂BNT体系能带结构由间接带隙转变为直接带隙,出现由Mo的4d轨道所贡献的杂质能级,导致带隙减小。Mo掺杂导致的杂质能级与莫斯-布尔斯坦效应之间存在带隙调控相互竞争关系,可有效调控BNT体系能带结构。
关键词: 钛酸铋钠, 第一性原理, 光学带隙, 能带结构, 态密度
0 引 言
随着环保意识的提高,人们迫切希望解决日益增长的能源需求与传统化石能源产生碳排放污染之间的矛盾,太阳能被认为是有前景之一的清洁能源[1],如何提高材料对太阳光吸收引起众多研究者关注[2]。 具有自发极化特点的铁电材料,由于所产生的铁电光伏效应跟传统 p-n 结的光伏响应不同,在光吸收应用中表现出很好的潜力[3],故研究人员一直在尝试通过各种途径改善铁电半导体材料的光学性能,其中离子掺杂调控是一种简单有效的方法[4]。
Na0. 5Bi0. 5TiO3 (BNT)基无铅陶瓷是一种绿色 ABO3 型钙钛矿铁电材料,具有一定的光吸收能力,但纯BNT 光学带隙较高(3 ~ 3. 2 eV)[5],故希望通过离子掺杂调控改善其光吸收强度,降低光学带隙,提高光吸收强度。 Gebhardt 等[6]研究发现过渡族阳离子会占据 B 位,导致氧八面体中心偏离,造成畸变,改变钙钛矿陶瓷的光学带隙。 Pham 等[7]研究发现,通过掺杂 Mo 可以使 NiTiO3 陶瓷光吸收强度增加,从而减小光学带隙值。 Liu 等[8]通过第一性原理计算研究发现,掺杂 Mo 可以改善 SrTiO3 光吸收波长范围。 Khan 等[9]、Yu等[10]通过理论计算研究发现,掺杂 Mo 可减小 TiO2 带隙,为 n 型掺杂。 但目前对通过 Mo 掺杂 BNT 基无铅铁电陶瓷进行带隙调控的研究较少,其对带隙的影响规律与机理有待深入研究,本文通过掺杂不同成分的Mo 研究了 BNT 基陶瓷的光吸收和光学带隙的变化,通过第一性原理计算分析了掺杂体系带隙变化的机理。
1 实验与计算方法
1. 1 样品制备与表征
采用分析纯的原料Bi2O3 、Na2CO3 、TiO2 和MoO3 , 其纯度均大于99%(质量分数) , 根据Na0. 5Bi0. 5 (Ti1 - xMox)O3 (BNT-Mox,x = 0% ,0. 5% ,1. 0% ,1. 5% ,2. 0% )准确称量配料后,以无水乙醇为球磨介质球磨 24 h 后置于 80 ℃烘干,采用两步过筛法处理粉料。 第一步过筛使用 100 目(160 μm)的筛网,将烘干后的粉料通过筛网过滤,然后将过滤后的粉料放入预烧炉,在 850 ℃预烧环境下保温 2 h,将预烧完成的粉料破碎后加入 7% 的 PVA 黏结剂适量,搅拌均匀后再次烘干,重新研磨成粉料。 第二步过筛使用 150 目(100 μm)的筛网,将研磨后的粉料过滤。 粉料经过两步过筛后,称取适量粉料放入压片模具进行压制成型,在 10 MPa 压力下将粉料压制成直径 19. 5 mm、厚 1. 2 mm 左右的圆坯。 将坯料置于马弗炉中慢速升温至600 ℃后保温 2 h 排胶,然后升温至 1 120 ℃保温 3 h。 由于烧制过程中会造成 Na 和 Bi 的挥发,故采用埋烧法将原粉料覆盖在样品表面后烧制,以减少样品损失。
测试使用德国 Bruker D8-2-Advance 型号的 X 射线衍射仪表征陶瓷样品的晶体结构,测试采用 Cu 靶,Kα射线,测试范围为 20° ~ 80°, 步长为 0. 02°, 扫描速度为 8 (°) / min。 采用法国 HORIBA LabRAM HREvolution 高分辨显微共焦拉曼光谱仪研究样品的价键振动变化,室温下采用 Ar 532 nm 离子激光,设置测试范围为 40 ~ 1 000 cm-1,单次采谱时间为 5 s,循环次数为 2,实时采集曝光时间为 2 s。 使用美国PerkinElemer Lamber750S 紫外-可见-近红外分光光度计研究样品的光吸收,测量波长范围为 200 ~ 1 000 nm,间隔点设置为 5 nm。
1. 2 模型构建与计算方法
室温下 BNT 属于三方相[11]
,三方相 BNT 空间群为 R3c,如图 1(a)所示。 晶格常数 a = b = c = 0. 55 nm,α = β = γ = 59. 80°[12],使用 VESTA(visualization for electronic and structural analysis)[13]建立 2 × 2 × 2 超晶胞,采用原子替代的掺杂方法,用 Mo 替代 Ti 原子[14],如图 1(b)所示。
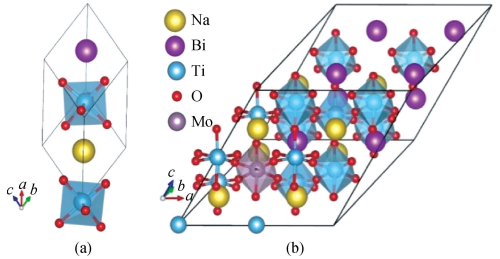
图 1 (a)空间群为 R3c 的纯 BNT;(b)BNT 的 2 × 2 × 2 超晶胞掺 Mo
计算使用 VASP 软件包(the vienna Ab initio simulation package)[15-17]对模型进行结构优化和计算。 计算采用的交换关联泛函为局域密度近似(local-density approximation, LDA)[18],采用的赝势方法为投影缀加平面波赝势 ( PAW) 方法[19-20]。 计算考虑的价电子为 Bi:5d106s26p3、Na:2p63s1、Ti:3p63d24s2、O:2s22p4、Mo:4d55s1。 由于计算中含有过渡族金属元素,而过渡族金属元素的电子间库伦相互作用不可忽略,为强关联电子体系,故采用在位库伦修正的方法( on-site coulomb correction),即 LDA + U 计算法(Ti:Ueff = U - J =5. 8 eV;Mo:Ueff = U - J = 2 eV)[14]。 计算使用 Monkhorst-Pack 方法设置 k 点网格为 4 × 4 × 4,设置平面截断能 Ecut = 500 eV,原子间相互作用力不大于 0. 2 eV/ nm,能量收敛精度为 1 × 10-5eV。
2 结果与讨论
图 2 为 BNT-Mox 陶瓷的 XRD 谱。 由图 2 ( a) 可看出,所有样品均为钙钛矿结构且无明显杂峰。 从图 2(b)看出随着 Mo 掺杂量的增加,在 40. 4°处的(210)峰,峰宽变窄,46. 9°处(220)峰的峰宽和峰强有劈裂变化,样品掺杂前后从三方相转变到伪立方相[21]。
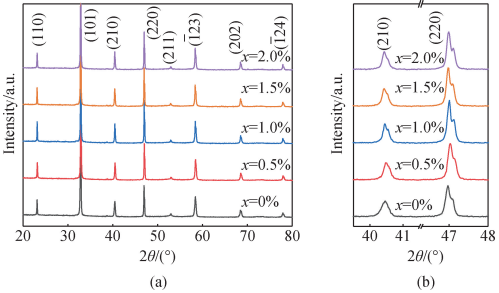
图 2 BNT-Mox 陶瓷的 XRD 谱
图 3 为室温下 BNT-Mox 陶瓷的拉曼散射光谱,与已报道的 BNT 基陶瓷拉曼光谱类似[5,22]。其各个峰的位置分别位于 56 cm-1、123 cm-1、274 cm-1、517 cm-1、581 cm-1和 768 cm-1,其中 40 ~ 200 cm-1是对应 A 位阳离子变化有关的范围[23];200 ~450 cm-1是对应 Ti—O键变化有关的范围;450 ~ 700 cm-1是对应 B 位氧八面体变化有关的范围。700 cm-1 以后是对应A1(longitudinal optics, LO)与 E(LO)两种模式重叠的范围[24-26],在 768 cm-1附近为受 TiO6 八面体团簇扭转位置影响的 LO3 模式[27-29]。 可以看出,随着掺杂量的增加,在 274 cm-1和 517 cm-1处峰强增大,表明 Mo 掺杂影响了 Ti—O 键与 TiO6 八面体,进入了 B 位。
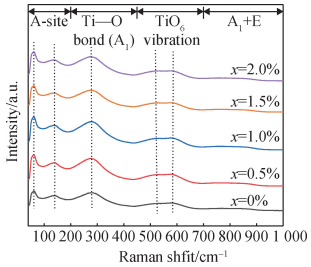
图 3 BNT-Mox 拉曼散射光谱
图 4(a)为 BNT-Mox 的紫外-可见-近红外光谱法测定的吸收光谱,其测量波长范围为 200 ~ 1 000 nm。可看出样品的光吸收主要集中在 300 ~ 400 nm 波长范围内,Mo 掺杂后吸收强度有显著提升,且当 x = 1. 0%时吸收强度最大值为 69% 。 光吸收强度与光学带隙之间有相关性,Huang 等[30]认为带隙减小是因为 Mo 离子掺杂影响 Ti 离子轨道导致的。 图 4(b)为样品光学带隙随掺杂成分的变化,可由 Kubelka-Munk 函数曲线的切线在横坐标上的截距得出[31]。 可看出随着掺杂量的增加,光学带隙先减小再增大,当 x = 1. 0% 时达到最小值2. 33 eV,随后增大至 2. 46 eV。
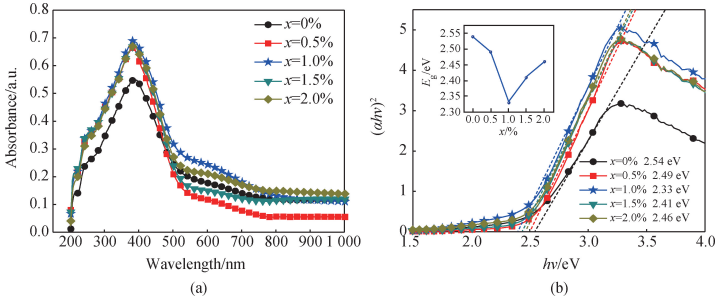
图 4 (a)掺杂量对 BNT-Mox 陶瓷的光吸收谱的影响;(b)掺杂量对 BNT-Mox 陶瓷光学带隙图影响
为了进一步研究 Mo 掺杂 BNT 可以增强光吸收和调控光学带隙的机理,利用 VASP 和 VASPKIT 后处理软件包,结合 VESTA 等建模软件[13,17],建立纯 BNT 与 Mo 掺杂 BNT 的模型,并分别计算和处理得出二者的能带结构与总/ 分态密度图。 图 5(a)为纯 BNT 的能带结构图,其中能带的路径和高对称点由 Seekpath 等[32]以及 Setyawa 等[33]研究得出。 高对称点处的竖线表示该点有不同的路径。 由图 5(a)可看出,价带顶位于 Γ点处,导带底位于 T 点处,为间接带隙,与周树兰等[34]结论趋势一致。 图 5(b)为 BNT-Mox 的投影能带图,其中红色竖线(彩色效果详见电子版)标记为 Mo 元素投影,标记越大表示所占权重越大,可以看出 Mo 掺杂主要对体系的导带产生影响。 Mo 掺杂后相对于纯 BNT 导带降低,且在费米能级处有能带穿过,表明在掺杂体系禁带内有杂质能级[10,35]。 导带底与价带顶在同一 Γ 点处,为直接带隙。
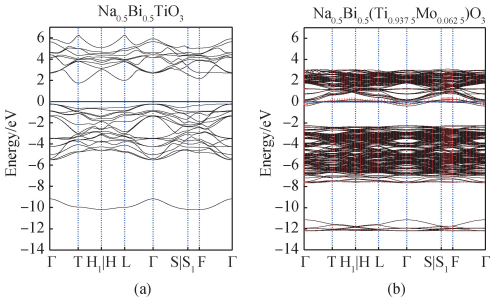
图 5 (a)纯 BNT 能带结构图;(b)Mo 掺杂 BNT 能带结构图
图 6(a)为纯 BNT 的总态密度(TDOS)与各元素分波态密度(PDOS)图,可得价带的主要贡献为 O 2p 轨道,导带的主要贡献为 Ti 3d 轨道。 由图 6(b)态密度图可知,掺杂体系内的杂质能级主要由 Mo 4d 轨道贡献,费米能级处的导带主要由 Mo 4d、Ti 3d 与 Bi 6p 轨道杂化形成,价带主要贡献为 O 2p 轨道。 综上可知:当 Mo 掺杂量低时,能带结构由间接带隙转变为直接带隙,引入的杂质能级起主导作用,导致本征带隙减小;随着 Mo 掺杂量进一步增加,由于费米能级进入导带中,导带底的能量状态被占据,导带内电子占据态将费米能级推向更高的能量,掺杂量高时莫斯-布尔斯坦[36-37]效应起到主导作用,可引发表观光学带隙增加[38-39];这两种带隙调控机制相互竞争,其主导作用随 Mo 掺杂量增加而改变,从而导致光学带隙先减小后增大。
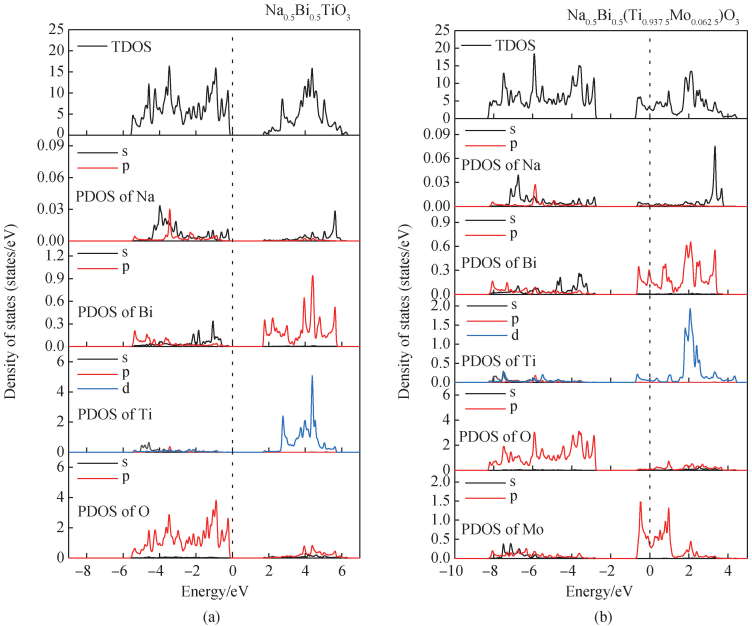
图 6 (a)纯 BNT 的 TDOS 与 PDOS 图;(b)Mo 掺杂 BNT 的 TDOS 与 PDOS 图
3 结 论
(1)本文采用传统固相法合成了 BNT-Mox 陶瓷,XRD 和拉曼光谱表明 BNT 陶瓷掺杂过程中发生从三方相到伪立方相的转变。
(2)随着 Mo 掺杂量的增加,光学带隙先减小后增大,在 Mo 掺杂量为 x = 1. 0% 时,光吸收强度达到最大值,光学带隙值达到最小值 2. 33 eV。
(3)通过理论计算分析,Mo 掺杂 BNT 导致体系禁带内有杂质能级,杂质能级主要由 Mo 4d 轨道贡献,导致能带结构由间接带隙转变为直接带隙,本征带隙减小。 Mo 掺杂导致的杂质能级与莫斯-布尔斯坦效应之间存在带隙调控相互竞争关系,可有效调控 BNT 体系能带结构。
参考文献:略
声明:本文由 CERADIR 先进陶瓷在线平台的入驻企业/个人提供或自网络获取,文章内容仅代表作者本人,不代表本网站及 CERADIR 立场,本站不对文章内容真实性、准确性等负责,尤其不对文中产品有关功能性、效果等提供担保。本站提醒读者,文章仅供学习参考,不构成任何投资及应用建议。如需转载,请联系原作者。如涉及作品内容、版权和其它问题,请与我们联系,我们将在第一时间处理!本站拥有对此声明的最终解释权。


{kind=link}
{kind=link}