此文为纯英文版,阅读请切换英文版浏览
声明:本文由 CERADIR 先进陶瓷在线平台的入驻企业/个人提供或自网络获取,文章内容仅代表作者本人,不代表本网站及 CERADIR 立场,本站不对文章内容真实性、准确性等负责,尤其不对文中产品有关功能性、效果等提供担保。本站提醒读者,文章仅供学习参考,不构成任何投资及应用建议。如需转载,请联系原作者。如涉及作品内容、版权和其它问题,请与我们联系,我们将在第一时间处理!本站拥有对此声明的最终解释权。
此文为纯英文版,阅读请切换英文版浏览
声明:本文由 CERADIR 先进陶瓷在线平台的入驻企业/个人提供或自网络获取,文章内容仅代表作者本人,不代表本网站及 CERADIR 立场,本站不对文章内容真实性、准确性等负责,尤其不对文中产品有关功能性、效果等提供担保。本站提醒读者,文章仅供学习参考,不构成任何投资及应用建议。如需转载,请联系原作者。如涉及作品内容、版权和其它问题,请与我们联系,我们将在第一时间处理!本站拥有对此声明的最终解释权。
CERADIR 将会持续更新我们的内容,如果你有任何疑问或建议,请联系我们!

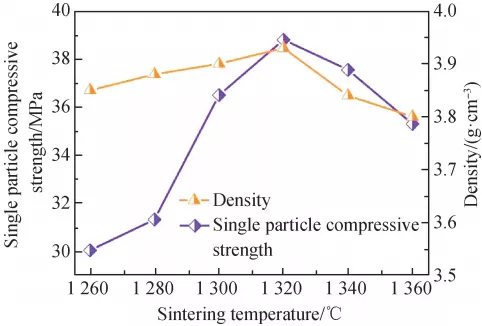
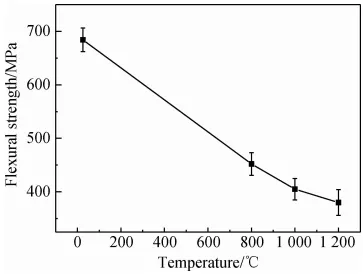

{kind=link}
{kind=link}